Comparative Genomics
比較ゲノム解析ページ > 納豆菌ゲノムプロジェクト納豆菌ゲノムプロジェクト
メンバー紹介
納豆菌ゲノムプロジェクトの榊原研究室メンバーを紹介します。 この他にも,慶應義塾大学先端生命科学研究所の板谷光泰教授, 成蹊大学理工学部情報科学科の長名保範助教, 国立遺伝学研究所の藤山秋佐夫教授,豊田敦特任準教授 も納豆菌ゲノムプロジェクトのメンバーでした。

西藤 ゆかり (OG)
納豆菌ゲノムプロジェクトの中心人物。WetからDryまで幅広く担当。

中村 祐太 (OB)
2010年度から納豆菌ゲノムプロジェクトに加入。納豆菌ゲノム解読後の新たなテーマの下,Wet実験を中心に担当。

クリス ポペンドフ (OB)
次世代シークエンサーデータの解析に貢献。 納豆菌ゲノムアッセンブリのためのプログラム開発を担当。

八谷 剛史 (OB)
納豆菌ゲノム解読後の解析からプロジェクトに加入。 納豆菌と近縁種の比較ゲノム解析を担当。Step 1:Wet実験 ゲノム抽出・サンプル調整
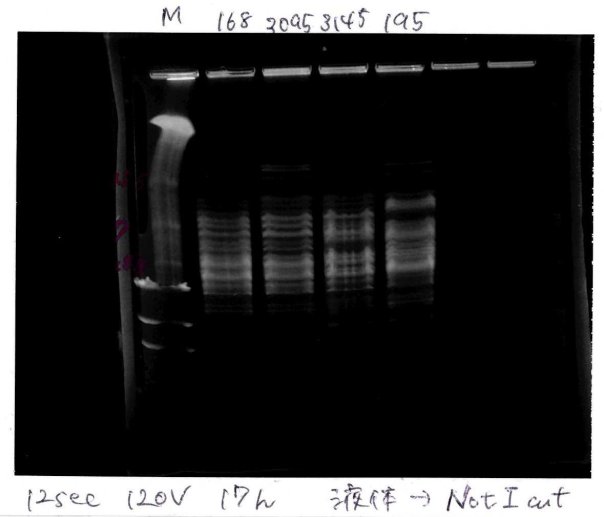
納豆菌ゲノムプロジェクトの最初のステップとして, 納豆菌からゲノムDNAを抽出しました。 共同研究をしている慶應義塾大学先端生命科学研究所 板谷光泰教授に納豆菌BEST195株を提供いただき、 板谷先生のいらっしゃる鶴岡の先端生命研にてゲノムDNA抽出法を教えていただきました。 現在は、榊原研究室内でもゲノムDNAの抽出ができるようになりました。
Step 2:Wet実験 次世代シークエンサー
2010年春,GenomeAnalyzer IIx (GAIIx) という機種の次世代シークエンサーが慶應大理工学部に導入されました。 次世代シークエンサーのシークエンシング性能は非常に高いですが, 一方で,使いこなすまでに試行錯誤の積み重ねが必要になります。 榊原研究室は GAIIx の主な利用者として, 次世代シークエンサーのメンテナンスやノウハウの蓄積を行っています。 2010年秋現在,慶應大理工学部の GAIIx は 1 Run 当たり 240 億塩基対 (ヒトゲノムの約 8 倍)のスループットを安定して実現できています。
納豆菌ゲノムプロジェクトを行ったときにはまだ理工学部に GAIIx が導入されていませんでしたので, 国立遺伝学研究所の次世代シークエンサーを使わせてもらいました。 GAIIxを動かすためには、たくさんの試薬を使ってサンプルを 調整する必要があります。 そのときには約 10 億塩基対の配列データが得られました。 納豆菌のゲノムサイズは約 410 万塩基対。 約 250 倍の配列データを得たことになります。 次世代シークエンサーで得られる配列データは, ゲノム配列中の 35-100 塩基対長の断片配列なので, このように小さな断片をつなぎ合わせてゲノム配列を解読するには, ゲノムサイズの何百倍もの配列データが必要になります。
Step 3:データ解析 短い断片配列のつなぎ合わせる
先ほど述べたとおり、 次世代シークエンサーでは大量の短い断片の配列を得ることができますが、 これらの短い断片配列のつなぎあわせは非常に困難です。 そこで、榊原研究室の計算機の力を活用します。 先行研究では、二つの手法が提案されていました。
①同じ種あるいは非常に近い種のゲノムが解読されている場合、 解読されている配列に次世代シークエンサーの出力であるリード配列をマッピングして、 違う部分を探す方法。
②リード配列同士の重なりの情報のみを用いてつなぎあわせて コンティグと呼ばれる配列を作る方法。
納豆菌の場合、近縁の枯草菌ゲノムは解読されていますが、 ①の方法では挿入や欠失を検出することができません。 一方②の方法を用いるだけでは、コンティグ配列同士をつなぎあわせることができず、 ばらばらのままになってしまいます。 そこで、今回は①と②の方法をあわせた新たな手法の確立を行い、 問題を解決していきました。
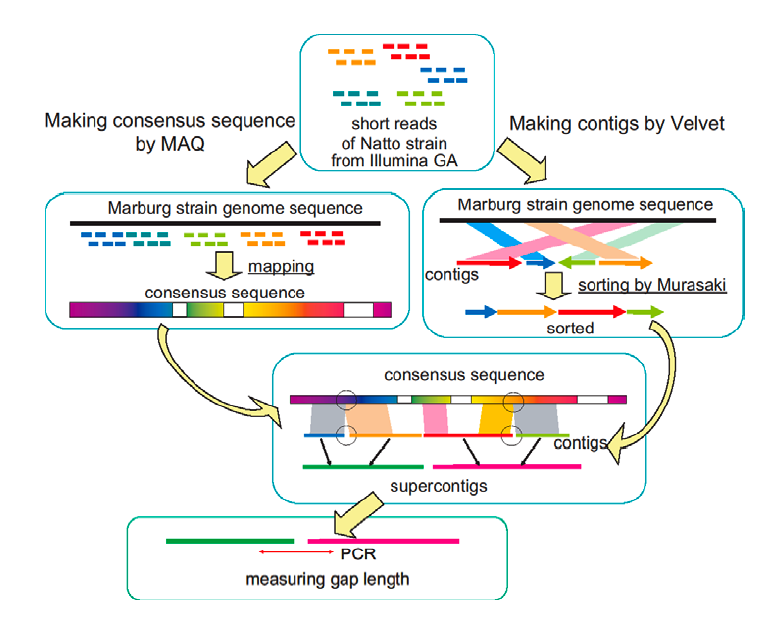
Step 4:Wet実験 コンティグ配列からゲノム配列へ
納豆菌は原核生物ですので,1本の染色体を持っています。 ゲノムプロジェクトの目標は染色体のゲノム配列を調べることですが, スタートが次世代シークエンサーのリード配列という非常に短い断片配列なので, コンティグを作ってもまだ完全な染色体配列にはなりません。 納豆菌ゲノムプロジェクトの場合,Step 3 の結果, 約20本程度のコンティグ配列が得られました。 Step 4 の目標は,この20本のコンティグ配列を並び替えて, 1本の染色体へと統合することです。
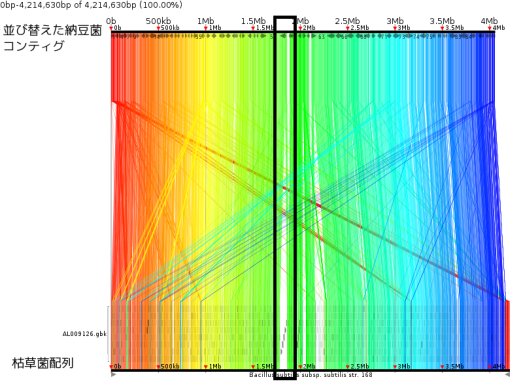
納豆菌ゲノムプロジェクトでは,まず,納豆菌の近縁種である枯草菌の情報を使って, 納豆菌コンティグの並びを予測しました。 その結果,納豆菌コンティグを並び替えると, 枯草菌ゲノムと非常に良く似ていることが分かりました(下図)。 このようにして並びを予測した後は, PCR法とキャピラリーシークエンサー(従来のシークエンサー)を用いて, 予測した並びが正しいかをWet実験によって確認しました。 榊原研究室での研究では, Wet実験とDry解析を交互に繰り返すと言う例もたくさんあります。
Step 5:データ解析 比較ゲノム解析
解読されたゲノム配列から生物学的な知見を導出するためのアプローチとして, 比較ゲノム解析は非常に有用です。 例えば,納豆菌ゲノムと枯草菌ゲノムを比較すると全体的には非常に良く似ているのですが, 部分的には,劇的に変化しているゲノム領域もあります。 納豆菌は納豆のねばねばを作ることができますが, 枯草菌は納豆のねばねばを作ることができません。 納豆菌ゲノムと枯草菌ゲノムを比較して異なっている領域は, 納豆のねばねばを作るために必要なゲノム領域だと考えられます。
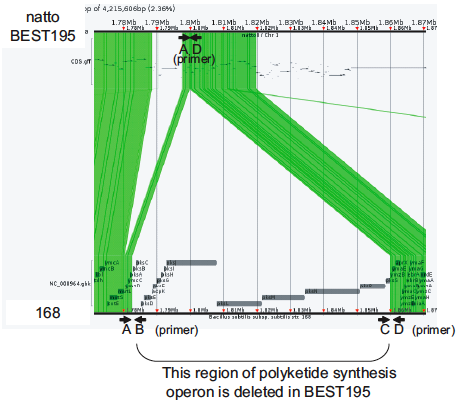
例えば,枯草菌はポリケチド合成に必要な遺伝子群を持っていますが, それらの遺伝子は納豆菌で欠失されていることが分かりました(下図)。 納豆のねばねばの正体はγグルタミン酸という化合物ですので, ポリケチド合成遺伝子群など二次代謝に関連する遺伝子は, ねばねば合成に関与する遺伝子として有用な候補と考えられます。 その他にも,二次代謝関連遺伝子の転写因子への変異や, 細胞間コミュニケーションに関わる遺伝子への変異など, ねばねば合成に関わる遺伝子の候補を多数見つけることができました。 今後の展望として,これらのねばねば合成遺伝子の候補を手がかりとして, ねばねば合成経路を解明してゆきます。